Our Company
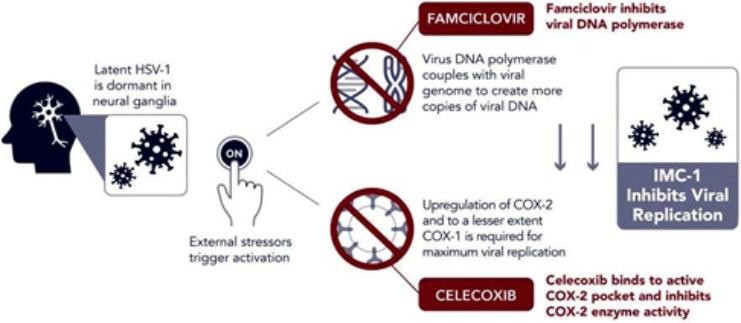
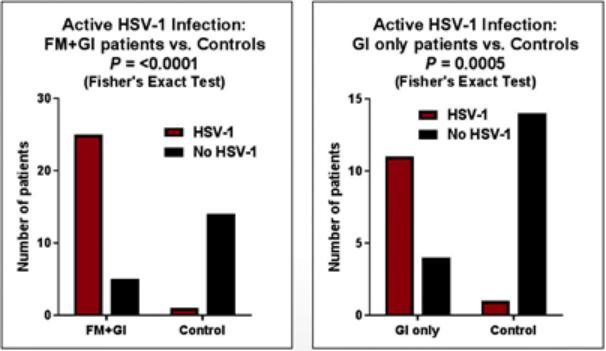
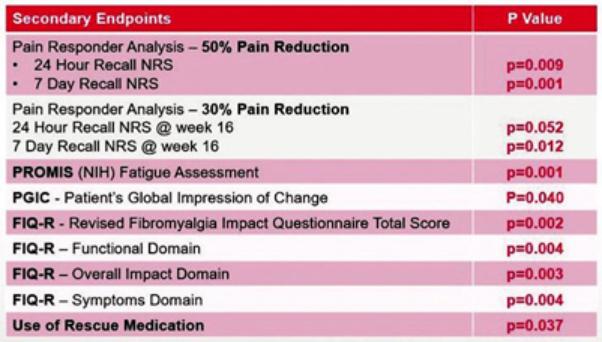
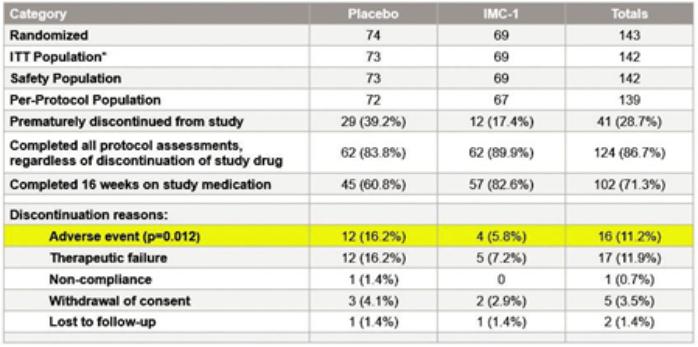
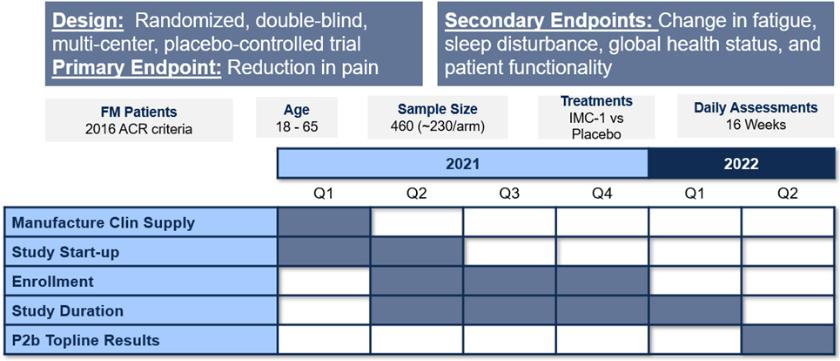
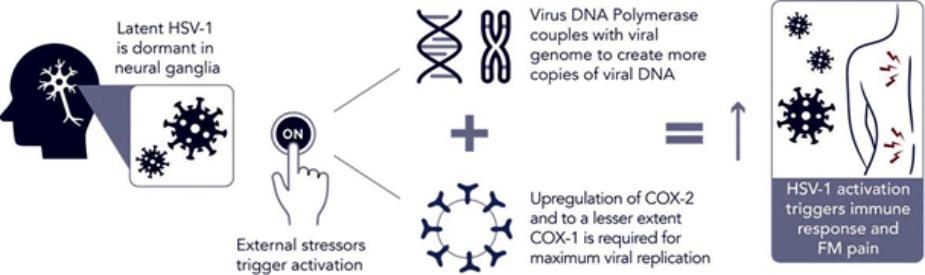
We are a development-stage biotechnology company focused on advancing novel antiviral therapies to treat diseases associated with a viral triggered abnormal immune response such as fibromyalgia (“FM”). Overactive immune response related to activation of tissue resident Herpes Simplex Virus-1 (“HSV-1”) has been postulated to be a potential root cause of chronic illnesses such as FM, irritable bowel disease (“IBS”), chronic fatigue syndrome and other functional somatic syndromes, all of which are characterized by a waxing and waning manifestation of disease. While not completely understood, there is general agreement in the medical community that activation of HSV-1 is triggered by some form of environmental and/or health stressor. Our lead candidate, IMC-1, is a novel, proprietary, fixed dose combination of famciclovir and celecoxib. IMC-1 represents a novel combination, dual mechanism antiviral therapy designed to synergistically suppress HSV-1 activation and replication, with the end goal of reducing viral mediated disease burden.
IMC-1 combines two specific mechanisms of action purposely designed to inhibit HSV-1 activation and replication, thereby keeping HSV-1 in a latent (dormant) state or “down-regulating” HSV-1 from a lytic (active) state back to latency. The famciclovir component of IMC-1 inhibits viral DNA replication. The celecoxib component of IMC-1 inhibits cyclooxegenase-2 (“COX-2”) and to a lesser degree COX-1 enzymes, used by HSV-1 to amplify or accelerate its own replication. We are unaware of any other antivirals in development for the treatment of FM. This novel approach was a germane consideration in FDA designating IMC-1 for fast-track review status for the treatment of FM. IMC-1 has also been granted a synergy patent based on the fact that neither of the individual components has proven effective in the management of fibromyalgia, yet the dual mechanism combination therapy generated a result in preliminary studies that appears to be greater than the sum of its parts.
Dormant HSV-1 is Reactivated by External Triggers and Amplifies
Its Own Replication via Cyclooxygenase (COX 1 and COX 2) Enzymes
6